By Zachary Coulson and James J. Dowling
Program for Genetics and Genome Biology, Hospital for Sick Children
Department of Molecular Genetics, University of Toronto
Publication (February 2022)
Rowel, K., Lim, Q., Yokota, T. et al.
Development of DG9 peptide-conjugated single- and multi-exon skipping therapies for the treatment of Duchenne muscular dystrophy
PNAS, 119(9), e2112546119 (2022). https://doi.org/10.1073/pnas.2112546119
https://www.pnas.org/doi/10.1073/pnas.2112546119
Significant progress has been made over the past several decades related to disease understanding and therapeutic strategies for the lethal X-linked muscle disease Duchenne Muscular Dystrophy (DMD). DMD arises from mutations in the dystrophin (DMD) gene; out-of-frame single or multi exon deletions are the primary genetic cause . One recent advancement in DMD therapeutics is the application of modified antisense oligonucleotides called phosphorodiamidate morpholino oligomers (PMOs) to promote skipping of exons in dystrophin in order to restore its reading frame. Four PMO based “exon skipping” ASOs have been approved by the food and drug administration (FDA), based on their ability to (modestly) restore DMD levels in patient in muscle. Each of these ASOs targets a single exon to restore dystrophin’s reading frame . These therapies, however, are limited both by their effectiveness in restoring DMD levels as well as by the prevalence of the individual mutation(s) they address.
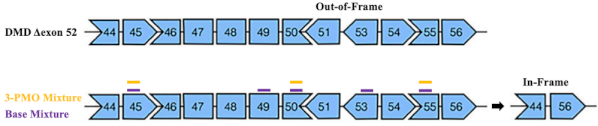
These limitations inspired Yokota and colleagues to evaluate multi-exon skipping as a therapeutic approach to skip a larger amount of the DMD gene in order to create for a more generalizable therapy. Their region of interest for multi-exon skipping targets exons 45-55, the deletion of which is an in-frame change that is associated with asymptomatic or mild clinical phenotypes . Targeting this region for multi-exon skipping has the potential to treat more than 40% of DMD patients. However, using PMOs to skip 10 exons simultaneously requires the use of 10 or more individual exon-targeting PMOs, resulting in decreased efficiency and a greater chance for off-target effects. This presented another set of limitations for Yokota and colleagues to overcome.
In this study, Yokota et al. attempted to create a minimized PMO mixture to see if they could achieve exon 45-55 skipping without having to target all 10 exons with 11 separate PMOs. They generated several “minimized” mixtures from their original pool of 11 exon 45-55 ASOs . Two mixtures were based on the most effective PMO derivatives from their total pool (called the “base” and “base -51” mixtures), and two were composed of PMOs targeting key exons in proposed DMD exon splicing groups (called the “block” and “3-PMO” mixtures) . These latter two mixtures were hypothesized to skip multiple exons by inhibiting a process known as back-splicing. In back-splicing, the 5’ end of a downstream exon is spliced to the 3’ end of an upstream exon to form circular RNA (circRNA), which may be composed of groups of multiple exons. Individual circRNAs undergo further splicing to remove intronic sequence, and are then spliced together with other circRNAs to form the mature mRNA transcript. By blocking pre-mRNA splicing at the 5’ end of a downstream exon and or the 3’ end of an upstream exon in a splicing group, the formation of a circRNA can be inhibited, thus resulting in the exclusion of multiple exons from the mature mRNA. Yokota et al. tested these minimized mixtures in various DMD patient cell lines and found that the “block” and “3-PMO” mixtures produced significantly stronger exon 45-55 skipping than their other minimized mixtures.
Having successfully developed a minimal cocktail of ASOs for exon 45-55 skipping, the authors then sought to address another limitation: permeability of PMOs within skeletal muscle. Several studies have addressed this by using peptide conjugated PMOs; however, these peptides can disrupt the cell membrane and are toxic at high concentrations . To overcome this, the authors tested a novel strategy of PMO conjugation to DG9. They examined this with an exon 51 targeting PMO in their humanized hDMDdel52;mdx mouse model. Unlike other peptide conjugates, DG9 has certain L-arginine residues converted to D-arginine to increase cell viability while lacking 6-aminohexanoic acid, which is known to cause toxicity . They found that single intravenous DG9-PMO treatment when compared to unmodified PMO produced 2.2- to 12.3-fold higher skipping and 1.5- to 3.4-fold higher DMD protein levels in skeletal muscles. Additionally, they found 14.4-fold higher skipping and 4.5-fold higher DMD protein levels in the heart. These findings were further supported with weekly treatments, which demonstrated (after 3 treatments) restoration of expression in skeletal muscle and heart to 2.8-3.9% and 7.7% of wild type (WT) respectively.
The authors then applied the DG9 modification to their “block” mixture comprised of 5 PMOs and tested exon skipping potential in the hDMDdel52;mdx mouse model. They found that local tibialis anterior (TA) intramuscular injections resulted in 9.5% exon 45-55 skipping efficiency compared to 15% using the “all” mixture (as measured in a different mdx model), and restoration of DMD protein expression to 0.76% WT levels compared to 0.12% WT levels in contralateral un-injected TA and 0.46% WT levels in saline injected controls.
In conclusion, the authors were able to achieve multi-exon skipping and increased DMD expression using a DG9 conjugated, minimized PMO mixture. The most exciting element of this study was the ability to fully skip exons 45-55 using only 5 ASOs, opening up the potential for multi exon skipping as a therapeutic strategy for up to 40% of DMD patients. However, even with the DG9 modification, the extent of full region skipping was quite modest, and the restoration of DMD protein with IM injection was less than 1%. This efficiency is even lower that what the authors achieved with their 11 PMO mixture, and is also lower than what has been reported with the existing single exon skippers (which themselves are considered only modestly impactful in terms of DMD protein restoration). There therefore remains important considerations related to optimization before this exciting potential strategy of multi exon skipping would be ready for clinical translation.
Figure 1. Cartoon adapted from Yokota et al. (2022) depicting the common DMD out-of-frame exon 52 deletion and PMO target locations for multi-exon skipping. Targeting exons 45, 49, 50, and 55 with the “base mixture” and exons 45, 50, and 55 with the “3-PMO mixture” results in exon 45-55 skipping producing an in-frame truncated DMD transcript.
At the time the above study was conducted, Kenji Lim was a Ph.D. Medical Genetics student at the University of Alberta, where he helped develop antisense oligonucleotide therapies for Duchenne muscular dystrophy and facioscapulohumeral muscular dystrophy. He has since finished his Ph.D. and have moved to the USA to pursue further training. He is currently a postdoctoral researcher (Banting Postdoctoral Fellow) at the Washington University in St. Louis, where he is now focused on understanding the very, very interesting roles of the innate immune system in heart failure.
Zachary Coulson is a master’s student at the University of Toronto in the Department of Molecular Genetics. He works in Dr. Jim Dowling’s lab at the Hospital for Sick Children and the University of Toronto. Zachary’s research involves using PMOs to skip multiple exons comprising functional domains in the nebulin gene to remove disease causing variants and treat nebulin related nemaline myopathy (NM) in cell and mouse models of NM.
Jim Dowling is clinician scientist pediatric neuromuscular specialist at the Hospital for Sick Children in Toronto. His work is focused on expanding disease understanding and developing and translating therapies for congenital myopathies.
This article is presented by the
Publication Highlights Committee.
Published on 19 May 2022.

16 Jan 2024 Anti-cN1A antibodies: epiphenomenon or pathogenic?

15 Nov 2023 Antisense oligonucleotide targeting DMPK in patients with myotonic dystrophy type 1: a multicentre, randomised, dose-escalation, placebo-controlled, p

15 Nov 2023 Mass spectrometry-based identification of new anti-Ly and known antisynthetase autoantibodies